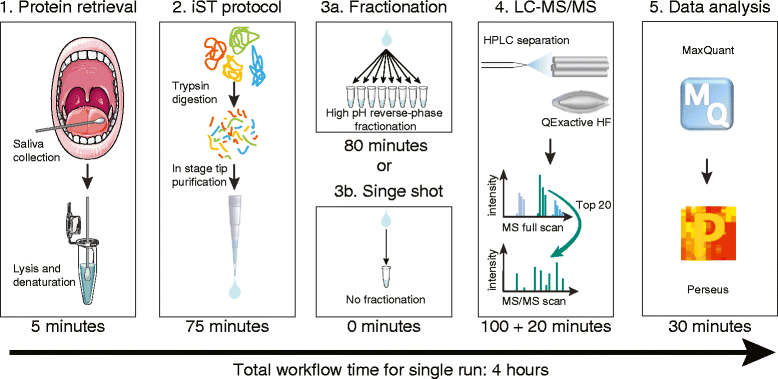
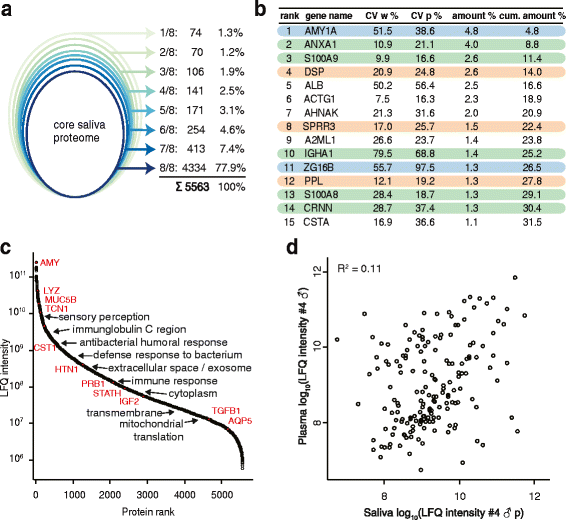
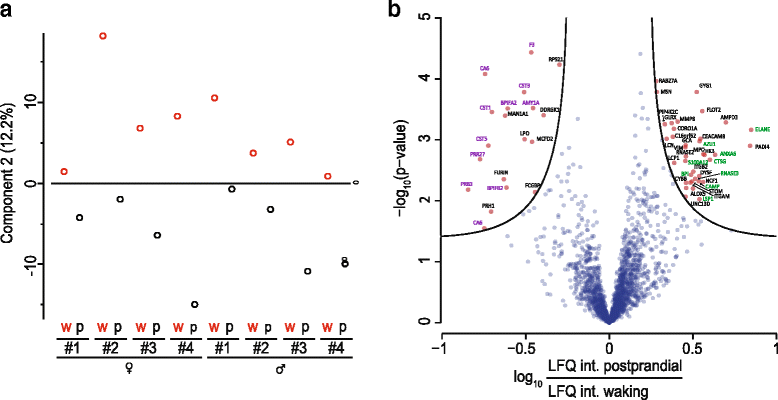
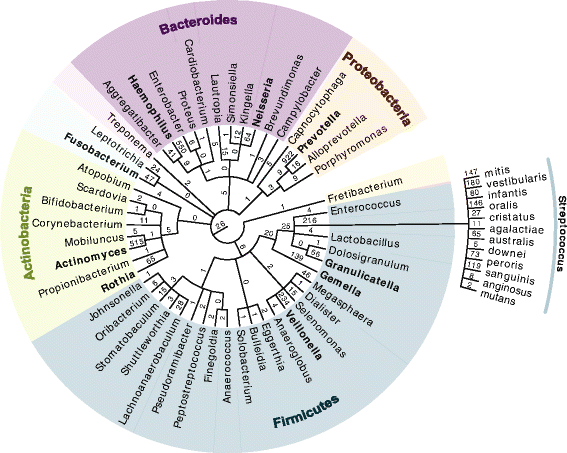
The application of
proteomics, or the analysis of proteins, to archaeology, is a fairly recent phenomenon – it only became viable thanks to developments in high-throughput, high-resolution tandem
mass spectrometry – and archaeological scientists are only just beginning to scratch the surface of the many ways in which this
technique might be used. Its potential is exciting, however.
One method, developed by Dr. Michael Buckley and Professor Matthew Collins at the University of York (Dr. Buckley is now at the University of Manchester), is called ‘ZooArchaeology by Mass Spectrometry’, or ZooMS for short. It uses
proteins from small fragments of bone, ivory, or skin (including artifacts made from these materials) to identify animal species that might otherwise be indistinguishable: for example, the identification of the animals whose hide was used to make the York Gospels parchment (see
CA 333).
Proteomics is also starting to be used to assess the contents of pots and other food vessels through the ‘residue’ left behind. While other studies, such as those carried out on the Chiseldon cauldrons (
CA 214) and pots from Durrington Walls (
CA 334) used lipid analysis, proteomics could potentially offer more specific results by being able to identify different parts of animals and plants, distinguishing between milk proteins, muscle proteins, structural proteins (namely, skin and bone), and keratins (hair and skin) from animals or seed proteins and root proteins from plants.
Slightly less well developed than ZooMS (which is quickly becoming an established method), but at a similar stage as food-residue analysis, is the use of proteomics to assess the composition of dental calculus. As emphasized in the previous issue’s ‘Science Notes’, teeth are one of the most informative parts of the human body, as they are relatively impervious to contamination and hold a plethora of clues about an individual regarding their diet, childhood origins, biological sex, and so on. But it is not just the teeth themselves that may be hugely helpful in reconstructing a person’s past: this new area of protein research is showing that the dental calculus – hardened, mineralized plaque – that forms on the surface of teeth, and the
proteins found within it, may be equally important.
Based largely on an individual’s health, oral hygiene, and salivary composition, dental calculus can be found on most archaeological remains to a varying degree, from a small amount of calculus seen around the gum lines to a large amount present around the majority of the tooth. This calculus is like a time capsule, preserving oral bacteria, host DNA, and consumed food and inhaled micro-debris. Its analysis opens up new ways of studying ancient human microbiomes, dietary practices, genetics, immune response, pathogenic presence, and general health.
The technology to do this kind of analysis already exists and several projects have now successfully recovered ancient proteins from samples of dental calculus. These early studies have mainly set out to test the waters though, identifying individual aspects of the oral microbiome and showing that it is more than possible to find pathogens, DNA, and food molecules. For example, one avenue of research currently being investigated is the identification of milk proteins, which are robustly preserved in dental calculus, to assess the origins of dairy production and consumption.
The problem comes, however, in determining the standards for such analysis, particularly in terms of how much the quantity and/or quality of protein information varies between samples, as well as between sites. Without such information, it is impossible to use the sequenced proteins comparatively, meaning that while we might be able to say which proteins are present in the dental calculus of an individual, we are not yet able to say much about what this may mean in terms of health and/or diet at the level of a population.
Recent experiments by Dr. Camilla Speller and a team from the University of York have examined the degree of variability between proteins able to be extracted from dental calculus. The results showed that there is substantial variation between individuals, but whether this is due to actual differences in the oral biome of each individual, the location of the calculus within the mouth, or degradation of the calculus over time is currently unknown. Additionally, while the researchers found that individual variability outweighed any inter-site differences, temperature and climate could still have their own effect on which proteins remain preserved.
To make this a more applicable technique, the team’s recent paper in Science and Technology of Archaeological Research stresses that, ‘more systematic studies exploring the quantity and quality of proteins preserved in both modern calculus, as well as a range of archaeological time periods, climatic zones, and burial environments, are required to differentiate and better understand individual variation and preservational biases’. Once the impact of these factors is fully appreciated, protein analysis of dental calculus could become one of the most informative types of analysis that can be done on human remains.
Reference: https://www.archaeology.co.uk/articles/pondering-the-possibilities-of-proteomics.htm