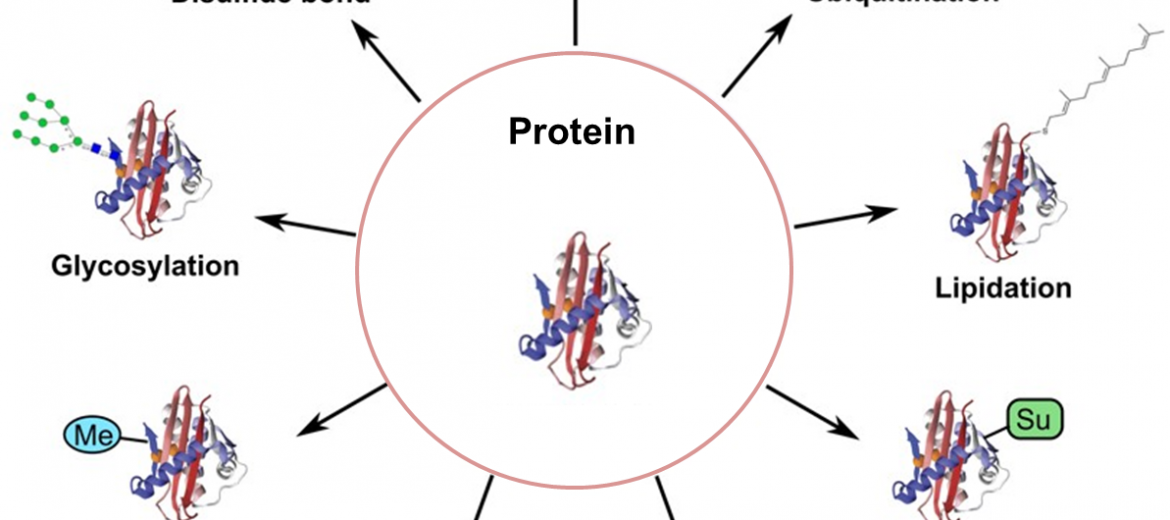
Strategies for Post-Translational Modifications (PTMs)
Post-translational modification (PTM) refers to the modification that occurs on a protein after translation catalyzed by enzymes. There are various types of PTMs, including phosphorylation, acetylation, methylation, glycosylation, and so on. PTMs play important roles in cell biology, including cell signaling, cell structure, DNA modification, and so on.
For the analysis of protein modification, traditional strategies, like radioactive labeling and Western blotting, can be specific and relatively quantitative, but they require prior knowledge of the modification type and are limited by antibody availability and specificity. Mass spectrometry (MS) is the suitable method for the analysis of protein modifications because it can provide universal information about protein modifications without a priori knowledge and locating the sites of modification. There are several strategies for PTM identification, including bottom-up, top-down, and middle-down approaches.
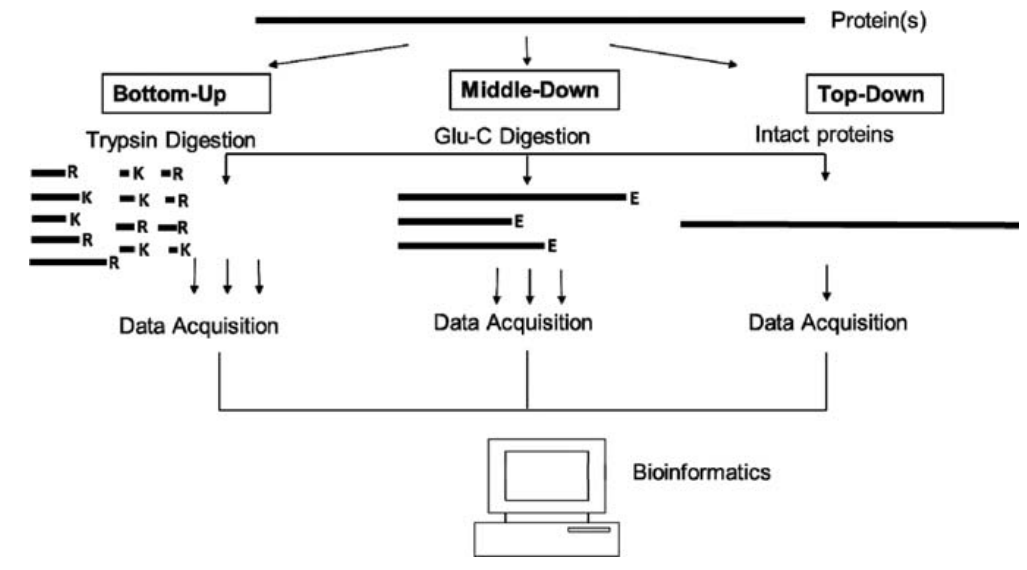
Figure 1. Bottom-up, middle-down, and top-down strategies (Moradian, Annie, et al, 2014)
Bottom-up strategy
The bottom-up strategy for PTM identification is the traditional proteomic approach. In bottom-up strategy, a protein is typically digested with an enzyme (like trypsin) into small peptides in gel or in solution. These peptides will be detected by mass spectrometer and modification can be mapped in the recovered peptides. The bottom-up strategy has the higher sensitivity than the top-down method. However, there are some intrinsic limitations in characterizing protein modifications for the bottom-up strategies. The first one is low percentage coverage of the protein sequence, which leads to the modification status of the unrecovered sequence portion remains unknown. In addition, the overall complexity of the sample is increased due to plentiful small peptide components by protein digestion. Moreover, the connection between modifications on disparate portions of a protein can be lost because the typical peptides from tryptic digestion contain only ≈5 to 20 amino acids.
Top-down strategy
Top-down proteomics is becoming a powerful strategy for analysis of protein modifications. For the top-down strategy, the whole protein is directly analyzed in the MS without digestion, so the full information of the modification state can be revealed. It can universally detect all the existing modifications, including PTMs (ie, phosphorylation, proteolysis, and acetylation) and sequence variants (ie, mutants, alternatively spliced isoforms, and amino acid polymorphisms) simultaneously in 1 spectrum without a priori knowledge. In this strategy, the molecular weight of an intact protein is first measured and compares it with the calculated value based on the DNA-predicted protein sequence, which can reveal modifications in the protein sequence globally. A specifically modified form of interest can be directly isolated in the mass spectrometer and subsequently fragmented by tandem MS for reliable mapping of the modification sites. Although top-down strategy has many advantages for protein modification analysis, there are still some challenges needed to be resolved, including protein solubility, sensitivity and detection limit, as well as the requirement of high-end instruments.
Middle-down strategy
Middle-down proteomics has recently emerged as high throughput strategy to define PTM co-existence frequency. The middle-down approach can be considered as a variant of the top-down approach. In this strategy, the proteins are needed to soft proteolysis to obtain the large peptides usually by using AspN and GluC. The peptides are then sequenced by tandem mass spectrometry using technologies similar to top-down. For example, the middle-down strategy can be used for histone analysis, in which proteins are commonly digested into peptides in the 3 to 9 kDa range. The middle-down approaches largely preserve the combinatorial modifications of the histone tail, while approaching the sensitivity of the bottom-up approach.
Mass spectrometry fragmentation strategies
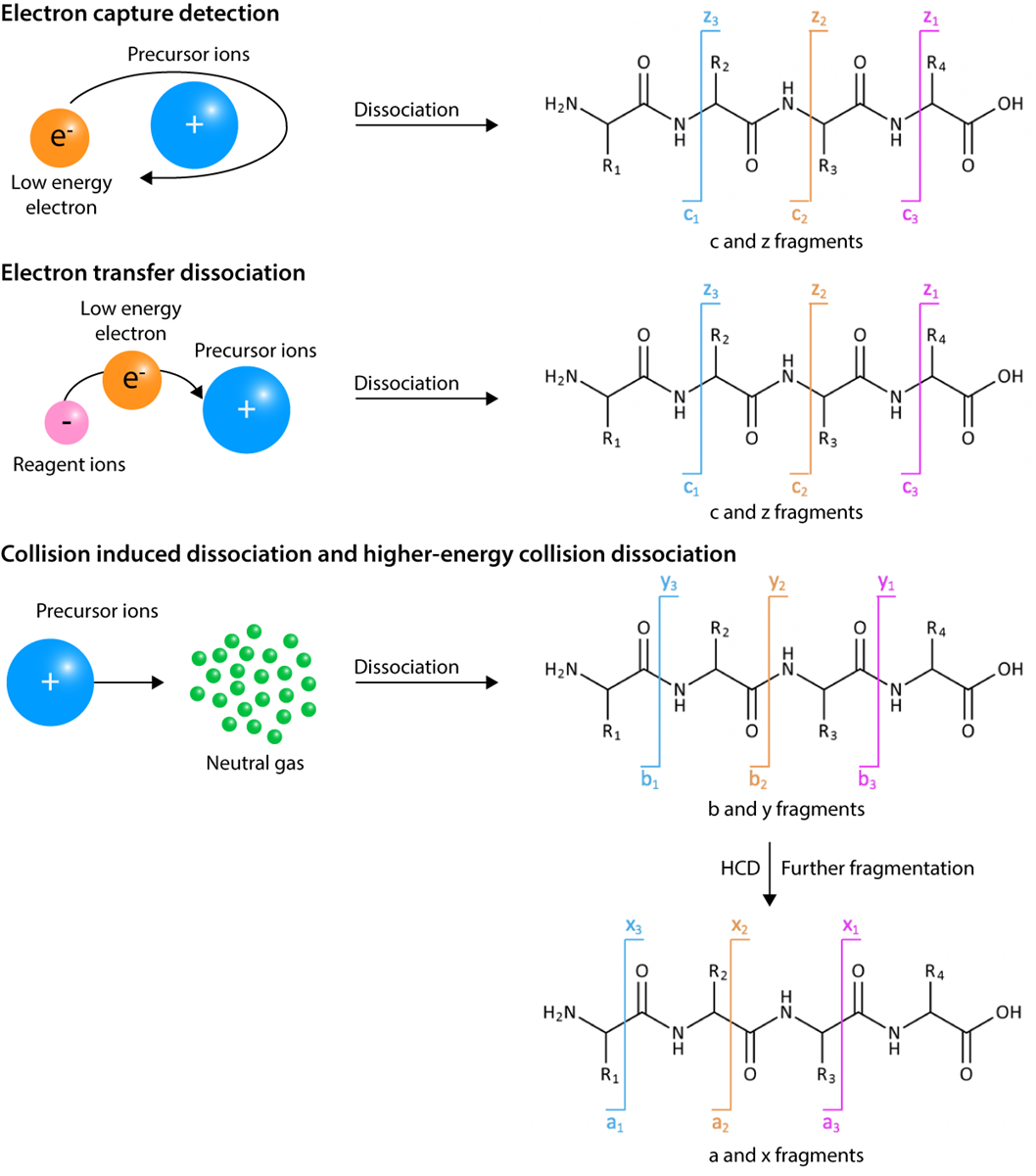
Figure 2. Mass spectrometry fragmentation methods
In MS-based PTM analysis, it is important to produce peptide fragmentation information for high confidence sequence identification and site localization of PTMs. There are several fragmentation strategies available, including collision induced dissociation (CID), higher energy collisional dissociation (HCD), electron capture dissociation (ECD), and electron transfer dissociation (ETD).
CID, also known as collisionally activated dissociation (CAD), is the most common and widely used unimolecular dissociation method. In CID, ions collide with neutral molecules (eg.) to be positively charged. The CID technique is generally more effective for small and low-charge state peptides. HCD, known as the beam-type CID, is a CID technique specific to the Orbitrap mass analyzer. Compared to CID, HCD have the higher activation energy and mainly generate y-type fragment ions. ECD and ETD, electron-based fragmentation methods, achieve fragmentation through neutralization of backbone protonation sites with thermal electrons (ECD) or radical anions (ETD). In this way, they can generate c and z- type fragment ions without losing the PTM localization information. CID can only be implemented on Fourier transform ion cyclotron resonance (FTICR) MS, ETD can be implemented on high-resolution tandem MS instruments. ECD and ETD perform better with highly charged state analytes, while CID is more efficient with low-charge state peptides. However, ECD and ETD are more suitable for detecting unstable PTMs for the reason that peptide backbone fragmentation is virtually independent of the amino acid sequence, neutral losses are reduced, and O-GlcNAc elimination does not happen.
Reference: https://www.creative-proteomics.com/blog/index.php/strategies-for-post-translational-modifications-ptms/
No comments:
Post a Comment